
MULTIPLE COMPARISONS, CONTRASTS, AND POST HOC TESTS Don't those three things kind of overlap with one another? I mean, aren't post hoc tests also multiple comparisons? Sure they are. I just wanted to make sure I was catching everyone who is interested in one or another of these topics, regardless of what they decide to call it. When it comes to multiple comparisons, R is both rich and poor. The base packages are quite limited (but still offer some powerful choices). However, there are optional packages that can be downloaded from CRAN that will do just about anything any sane person might want. One that I can recommend is a package called "multcomp". In this tutorial, I'm going to stick to what's available in the base packages. Furthermore, in this tutorial I'm going to stick to looking at one main effect, as might result from a one-way ANOVA, and main effects from between groups designs at that. I think it's necessary to cover this material first, before more complex situations can be tackled. We will tackle those in the relevant tutorials on factorial ANOVA and repeated measures designs. A note: For some of the things in this tutorial to work, it is necessary
to have your contrasts for unordered factors set to "treatment contrasts",
which is the R default. You can check that as follows:
Post Hoc Tests Post hoc tests are done after an omnibus ANOVA has been completed, when there has been a significant effect, and now it is desired to see exactly which groups are significantly different. Statisticians thrive on post hoc tests. There are more of these things than you can shake a stick at! Or would even care to shake a stick at!! I'm going to discuss four: the Tukey HSD test, the Fisher LSD test (with a note on Dunnett's test), the Bonferroni test, and the Holm-Bonferroni test. Tukey HSD test The Tukey Honestly Significant Difference test was illustrated in the
Oneway Between Subjects ANOVA tutorial. I'll repeat
that example here. Briefly, we are testing six insect sprays, A through F, and
after we spray garden plots with them, we are counting how many bugs are left.
What you don't get in this output is the actual HSD value. You don't really
need it, unless your homework requires that you calculate it! Which would not
be difficult. It's half the width of any of those CIs. (Notice they are all
the same width.)
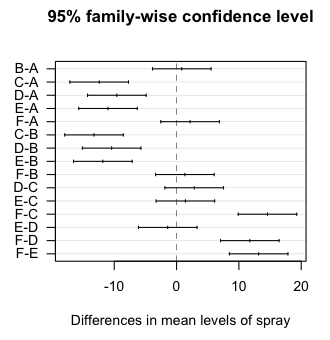
Also, the Tukey HSD test was originally intended for balanced designs only. The extention to unbalanced designs was developed by Kramer and has been incorporated into the R function. In the case of unbalanced designs, the proper name of the test is the Tukey-Kramer procedure. Fisher LSD test When I start talking about this test, my students immediately perk up, until I tell them that LSD stands for Least Significant Difference. The LSD test uses unadjusted p-values, and therefore it makes no attempt to control familywise error rate. If you use it as a protected test (i.e., only after seeing a significant effect in the omnibus ANOVA), it's good for about three or four comparisons without inflating the error rate. Beyond that, the inflation grows rapidly with increasing numbers of comparisons, if you're concerned about such a thing. It is among the most powerful of post hoc tests, however, and hey! Type II errors are errors too! Fisher LSD p-values for all possible pairwise comparisons can be obtained
this way.
How would we get the other comparisons? One way would be to calculate them by hand, which is not difficult using the information above. The LSD test is done as a t-test, with t equal to the difference in the means of the two groups that are being compared, divided by the standard error of that difference. The standard error is defined this way: Where n1 and n2 are the two group sizes, both 12 in this case, and s is the pooled standard deviation of the treatment groups, which can be obtained as follows.
> aov.out
Call:
aov(formula = count ~ spray, data = InsectSprays)
Terms:
spray Residuals
Sum of Squares 2668.833 1015.167
Deg. of Freedom 5 66
Residual standard error: 3.921902 # this is sqrt(1015.167/66) or sqrt of the MSE term
Estimated effects may be unbalanced
> 3.921902/sqrt(6) # calculation of se; /sqrt(6) being from sqrt(1/12 + 1/12)
[1] 1.60111
Notice the result matches that in the summary.lm table above. With a balanced
design, n1 and n2 are the same for all comparisons, and s is the same for all
comparisons, so the standard error will always be the same. Furthermore, the
difference in means between any two groups can be obtained from the "Estimates"
in the above table. So let's say we wanted the comparison between spray D and
spray E. We would do it this way.
> (-9.5833333 - (-11.0)) / 1.601110 # t [1] 0.8848029 > pt(0.8848029, df=66, lower=F) * 2 # two-tailed p-value (drop any sign on t) [1] 0.3794751Compare this to the p-value in the pairwise t-test table. When the design is unbalanced, the same standard error will not be used for
all tests in the summary.lm output. Neither will it be in the pairwise t-tests.
This is because n1 and n2 may vary between comparisons. Let's unbalance the
InsectSprays data, and see what effect that has. We'll
drop 5 cases selected at random.
Is there a way to get those other, non-A, comparisons? Other than doing them by hand, that is? Because hey! What do I own a computer for? The pairwise.t.test() function gives the p-values for them, but if you insist on having all the gory details, that's another matter. They can be had though if you're willing to do a little work. Let's say we
want all the B-comparisons. The reasons we got A-comparisons above is because
A is the first level of the spray factor.
The LSD (least significant difference) can be calculated this way.
What about confidence intervals? If you have LSD, a confidence interval is
just one more step. Let's do the 95%-CI for the D-E mean difference.
A note on Dunnett's test Notice above when you did summary.lm(aov.out), you got comparisons of group A with groups B, C, D, E, and F in turn. If group A is your control group, then these are Dunnett-like tests of all treatment groups with the control. The tests themselves are wrong, however, because Dunnett's adjustment to the p-value was not done. So far as I'm aware, there is no way to do it in the R base packages. There are optional packages that can be downloaded from CRAN that will do it, one of which is "multcomp". There are also tables of Dunnett's critical values online, for example, here. Bonferroni test The Bonferroni test is done exactly like the Fisher LSD test, but once the
p-values are obtained, they are multiplied by the number of comparisons being
made. In the case of InsectSprays, that would be 15 comparisons. The p-values
for all the comparisons can be obtained like this.
Holm-Bonferroni test The Holm-Bonferroni test is a very clever modification of the Bonferroni
correction. For details you'll have to consult a textbook (or Wikipedia), but
here is how you get the p-values.
Orthogonal Contrasts Clean up your workspace and search path. We're moving on to a new problem. We will look at a data set that records the dry-weight production of biomass
by plants as the result of three different treatments: a control, and two
experimental treatments.
Okay, stop! Don't go rushing headlong into the ANOVA without stopping and thinking first. What kind of hypotheses might we be interested in here? Well, any differences among the means. That's the traditional alternative for the omnibus ANOVA, but it's really kind of like beating the problem with a club! If we think about it, we might come up with hypotheses like these: (i) the treated groups will be different from the control group, and (ii) the two treated groups will be different from each other. (NOTE: Given that I don't know what the treatments are, I don't know if these are sensible hypotheses, but they COULD be. Use your imagination.) These are hypotheses which, like all hypotheses, we would want to express in advance. And now we want to come up with a method of data analysis that would allow us to test them. Enter what are called planned comparisons. The name makes perfect sense, since we're planning these comparisons in advance. We'll use contrasts to do the analysis. Contrasts do not have to be "orthogonal." Think of orthogonal as meaning something like "independent" or "unconfounded." I'll discuss nonorthogonal contrasts below. Right now, let's keep our contrasts orthogonal. There are several requirements for doing so. One has already been mentioned: a balanced design, which means orthogonal contrasts are done most often on experimental data (as opposed to quasi-experiments). Another one is that we can come up with a set of k-1 sensible orthogonal contrasts, where k is the number of levels of the IV we are testing. In this case, our IV has three levels, so we can have two contrasts, each corresponding to a sensible hypothesis. I think we have that. It now remains to code the contrasts. Each contrast is going to be coded with a contrast vector. Within the vector, each level of the IV is going to be given a code. Some of the codes will be positive, some will be negative. All of the positive codes in the vector should be the same, and they should add to +1. All of the negative codes in the vector should be the same, and they should add to -1. Thus, the codes in the vector should, overall, add to 0. The positively coded groups will be combined into one big "megagroup." The negatively coded groups will be combined into another big "megagroup." Then these two megagroups will be tested against each other, using what essentially amounts to a t-test. This is called a complex contrast, by the way, when you have groups combined into megagroups. A contrast can also just test one group against another, which is called a simple contrast. Fisher LSD tests are simple contrasts. Our first hypothesis suggests we want to lump the treatment groups into a megagroup, and test that megagroup against the control group. The contrast vector would be: These codes have to be in the SAME ORDER that R sees the levels of the variable, so we better check that. > levels(PlantGrowth$group) [1] "ctrl" "trt1" "trt2"Good! Our contrast will test "ctrl" (+1) against the megagroup "trt1+trt2" (each coded -1/2). "That's not going to give us anything," you're saying. "I've seen the means, and that's just not going to pan out!" Stop looking at the means! These are PLANNED in advance comparisons. You haven't got any means yet! For our second hypothesis, we want to compare "trt1" to "trt2". The secret here is to code any level not included in the contrast as 0. So the vector is: That will compare the second level of the IV to the third level, a simple contrast and, FYI, equivalent to a Fisher LSD test. By the way, it's completely arbitrary which levels are coded positive and which negative, but I'll give you a hint below as to what is a convenient way to choose.
> vec1 = c(1,-1/2,-1/2)
> vec2 = c(0,1,-1)
> ### now cbind the contrast vectors into the columns of matrix
> cons = cbind(vec1, vec2)
> cons
vec1 vec2
[1,] 1.0 0
[2,] -0.5 1
[3,] -0.5 -1
> ### it's helpful (but not really necessary) to name the rows of the matrix
> rownames(cons) = c("ctrl", "trt1", "trt2")
> cons
vec1 vec2
ctrl 1.0 0
trt1 -0.5 1
trt2 -0.5 -1
> ### to be orthogonal, all pairs of contrast vectors must have a dot product of 0
> ### here's a convenient way to test that for a contrast matrix of any size
> t(cons) %*% cons
vec1 vec2
vec1 1.5 0
vec2 0.0 2
> ### if this matrix multiplication results in a matrix that is nonzero ONLY on
> ### the main diagonal, then the contrasts are orthogonal
And we are coded! Now we assign that contrast matrix to the IV and run the
ANOVA. We're not really interested in the ANOVA itself, but this is how we're
going to get the contrasts.
> contrasts(PlantGrowth$group) = cons > aov.out = aov(weight ~ group, data=PlantGrowth)Now here's the tricky part. I'll do it, and then I'll explained what happened.
> summary(aov.out,
+ split=list(
+ group=list("ctrl v trts"=1, "trt1 v trt2"=2)
+ ))
Df Sum Sq Mean Sq F value Pr(>F)
group 2 3.766 1.883 4.846 0.01591 *
group: ctrl v trts 1 0.025 0.025 0.065 0.80086
group: trt1 v trt2 1 3.741 3.741 9.627 0.00446 **
Residuals 27 10.492 0.389
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
I get that wrong EVERY time! I always have to look at an example. We used the
usual extractor function, summary(aov.out), but
this time we used it with an option called split=.
The technical statistical jargon for what split does is "partition." Our
contrasts partitioned the treatment sum of squares. Because we had a full set
of orthogonal contrasts, the partitioning was spot on. I.e., the contrast SSs
add up to the treatment SS. What's up with that syntax?!
For once, it's actually quite sensible. Inside split we have a list that lists ALL of the factors we're doing contrasts on. Here we only have one, so our list exhausted itself at "group." Inside each of those factors we have another list. In that list, we give our contrasts sensible names, and we set those names equal to one of the columns in the contrast matrix (by number of the column). And that's pretty much it. "I thought you said they were like t-tests. That don't look like no t-test
to me!"
Here is another brief example. The data are based on an experiment by Eysenck (1974) on incidental learning, which is learning that takes place when one is not really attempting to learn but just through exposure to the material. Subjects were given a list of 30 words and one of five different instructions: count the letters in each word, think of a word that rhymes with each word, think of an adjective that could be used to modify the word in a sentence, form a mental image of the object described by the word, or a control condition in which the subjects were told to memorize the list. These are not the original data but were created to match the cell means and SDs. The data were taken from Howell's textbook (7th ed., 2010, Table 11.1, p. 319). The logic of the following contrasts is that only the control group was told
they'd be asked to recall the words, and that count and rhyme instructions
represent a shallow level of mental processing, while adjective and image
instructions represent deep mental processing. (Note: In the full experiment,
there were two age groups. These data are for the older subjects.)
Nonorthogonal Contrasts Those are not the contrasts I would have selected for the Eysenck study, and luckily for me, the statistics police will not come and put me in statistics prison if my contrasts are not orthogonal. (I'll probably get off with a warning.) There is no rule that says contrasts have to be orthogonal, even when the
design is balanced. Here are the contrasts I would test. (Note: Contrasts are
very personal things, kind of like underwear. People have different tastes.
As long as those tastes correspond to sensible hypotheses, the rule is live
and let live!)
Yeah, I know, it annoys me, too. So I've written a function to do it (see
Writing Your Own Functions and Scripts if you don't
know what I'm talking about). Open a script window (File > New Script in
Windows or File > New Document on a Mac), and copy and paste these lines into it.
If you'd rather report your results as t-tests, then t = sqrt(F) on dfe degrees of freedom, with a p-value as reported in the above table. These are two-tailed p-values when it comes to the t-test. For one-tailed, divide them in half, PROVIDED the effect went in the predicted direction. If you code the group(s) that you expect to have the larger mean with positive-valued contrast codes, then "diffs" will be positive if the effect went in the direction you expected. A Final Note Do planned contrasts require a p-adjustment for the number of comparisons? Oh no! You're not dragging me into that argument! Just try to hold it down to a reasonable number. created 2016 February 4 |